Metabolism
on-line - the virtual tutorial room
copyright © 2008 - 2015 David A Bender
An unusual cause of diabetes - how the pancreas senses a rise in blood glucose
There are two common types of diabetes mellitus
Type 1 diabetes (also known as insulin-dependent diabetes or juvenile onset diabetes)
Here the problem is one of failure to secrete adequate amounts of insulin, and patients are dependent on injection of insulin to maintain control over their blood glucose concentration. The condition usually develops in childhood and progresses with loss of pancreatic beta islet cells until there is almost no secretion of insulin.
Type 2 diabetes (also known as non-insulin-dependent diabetes or maturity onset diabetes)
Here the problem is one of loss of sensitivity of tissues to insulin action; the secretion of insulin may be normal or greater than normal. Although injection of insulin helps to maintain good control over blood glucose concentration, patients are not strictly dependent on an exogenous source of insulin. The problem usually develops in adult life and is commonly associated with obesity, especially abdominal obesity.
What are the main ways in which insulin acts to lower blood glucose?
Insulin stimulates uptake of glucose into muscle and adipose tissue. It achieves this by recruitment of glucose transporters from intracellular vesicles to the cell surface.
In the presence of insulin, glucose becomes major fuel for muscle contraction. There is increased synthesis of glycogen as a result of activation of glycogen synthase and inactivation of glycogen phosphorylase.
In adipose tissue, glucose is used for the synthesis of fatty acids and and the triacylglycerol for storage, as a result of increased activity of fatty acid synthase and decreased activity of hormone-sensitive lipase.
Liver uptake of glucose is not affected by insulin, but insulin stimulates the synthesis of glycogen from glucose, as in muscle, as a result of activation of glycogen synthase and inactivation of glycogen phosphorylase.
What are the main adverse effects of poor glycaemic control and hyperglycaemia?
Either failure of insulin secretion or decreased sensitivity of insulin receptors leads to elevated blood glucose (hyperglycaemia). Glucose reacts non-enzymically with amino groups in proteins, including:
collagen (hence increased arthritis and damage to epithelial basement membrane leading to damage to blood vessels, and contributing to circulatory problems, retinal damage and renal damage)
proteins in low density lipoprotein (hence increased risk of developing atherosclerosis)
crystallin in the lens of the eye ( hence the development of cataracts)
proteins in myelin (hence nerve damage)
haemoglobin – not a pathological problem, but diagnostically useful to monitor long-term control of blood glucose by measurement of glycated haemoglobin (haemoglobin A1c)
In addition, hyperglycaemia may lead to osmotic imbalance and dehydration. Glucose may also be reduced to sorbitol in nerve and other tissues, again leading to osmotic damage.
The beta-islet cells of the pancreas secrete insulin in response to an increase
in blood glucose. This problem is concerned with the way in which the beta-cells
detect the increased concentration of glucose.
One early hypothesis was that there is a glucose receptor on the outer surface of the beta -cell membrane, and when this binds glucose it initiates a series of intracellular events that lead to secretion of insulin.
Coore & Randle (1964) measured the secretion of insulin by rabbit pancreas
incubated in vitro with two concentrations of glucose, with and without the
addition of the 7-carbon sugar mannoheptulose, which they had previously shown
to be an inhibitor of phosphorylation of glucose to glucose 6-phosphate. Their
results are shown below.
Secretion of insulin (µg /minute /incubation) by rabbit pancreas incubated in vitro with 3.3 or 16.6mol /L glucose. (3.3 mmol /L is the glucose
concentration that would be seen in relatively prolonged fasting; 16.6 mol /L
is about the concentration that would be seen in the hepatic portal
vein after a moderately carbohydrate-rich meal):
| incubated with: | control |
+ mannoheptulose to inhibit phosphorylation
of glucose |
| 3.3 mmol /L glucose | 3.5 |
3.5 |
| 16.6 mmol /L glucose | 12.5 |
3.5 |
(From data reported by Coore HG & Randle PJ, Biochemical Journal 93: 66 – 77, 1964.)
What conclusions can you draw from these observations?
In the control incubations there is the expected increase in insulin secretion
in response to glucose; in the presence of mannoheptulose there is no increase
in insulin secretion in response to glucose. This suggests that the way in which
the pancreas senses increased blood glucose is not by binding to a receptor
on the cell surface, but by increased formation and metabolism of glucose 6-phosphate.
This problem concerns a small number of families with a clear pattern of dominant
inheritance of an unusual form of diabetes that develops in early childhood.
It is generally referred to as maturity-onset diabetes of the young (MODY),
although it is distinct from types 1 and 2 diabetes.
What is meant by a dominant pattern of inheritance?
Dominant inheritance means that the disease is manifest in heterozygotes, with
one normal and one abnormal gene. Commonly this is because the product of the
abnormal gene has undesirable effects, or because having only half the normal
amount of the enzyme or protein is not sufficient.
A recessive condition is where there must be two copies of the abnormal gene for the condition to be manifest because having only half the normal amount of enzyme does not have any effect.
As will be obvious by the time you have finished working through this problem,
MODY is a dominant condition because having only half the normal amount of an
enzyme does not permit adequate secretion of insulin in response to increasing
blood glucose.
Glucose enters the cells of most tissues by active transport, which is stimulated in response to insulin. In the absence of insulin, glucose transporters are sequestered in intracellular vesicles, which migrate to the cell surface and become active in the presence of insulin. This means that insulin promotes the uptake and utilisation of glucose in most tissues.
After entry, glucose is phosphorylated to glucose 6-phosphate before onward metabolism or use for synthesis of glycogen (in muscle) or fatty acids (in adipose tissue).
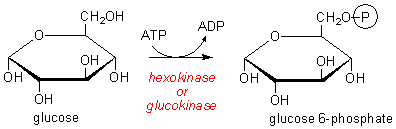
In the liver, glucose enters cells by passive diffusion and is then trapped
by phosphorylation to glucose 6-phosphate, which cannot cross cell membranes.
Glucose 6-phosphate is then either metabolised as a metabolic fuel or used to
synthesise glycogen and fatty acids.
Two isoenzymes catalyse the formation of glucose 6-phosphate from glucose
What is meant by the term "isoenzyme"?
Isoenzymes are enzymes that catalyse the same reaction, but encoded by different genes. They may differ in tissue or subcellullar distribution, reaction kinetics (i.e. a different value of Km), the range of substrate specificity or inhibitor sensitivity.
Two isoenzymes, hexokinase and glucokinase, catalyse the formation
of glucose 6-phosphate from glucose. Hexokinase has a broad specificity for
6-carbon (hexose) sugars, while glucokinase is more specific for glucose.
Km (mmol /L) |
expressed in: | |
| hexokinase | 0.15 |
all tissues |
| glucokinase | 8 |
liver and pancreatic islet cells, hypothalamus and enterocytes of the small intestine |
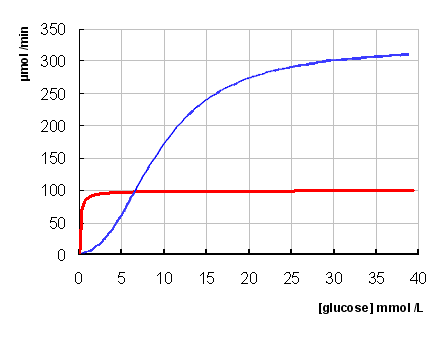 The normal range
of plasma glucose is between 3.5 - 5 mmol/L, rising in peripheral blood to 8
- 10 mmol/L after a moderately high intake of glucose. The concentration of
glucose in the portal blood, coming from the small intestine to the liver, may
be considerably higher than this after a moderately carbohydrate-rich meal.
The normal range
of plasma glucose is between 3.5 - 5 mmol/L, rising in peripheral blood to 8
- 10 mmol/L after a moderately high intake of glucose. The concentration of
glucose in the portal blood, coming from the small intestine to the liver, may
be considerably higher than this after a moderately carbohydrate-rich meal.
Note that glucokinase shows cooperativity in glucose binding, and has sigmoid
kinetics rather than hyperbolic, as for hexokinase.
What effect do you think changes in the plasma concentration of glucose will have on the rate of formation of glucose 6-phosphate catalysed by hexokinase in the liver?
Because its Km is so far below plasma concentration of glucose under all conditions, and glucose enters liver cells freely, hexokinase in the liver will always be acting at its Vmax – i.e. it will act at a constant rate, regardless of the concentration of glucose.
Note that in muscle and other tissues, glucose uptake is controlled by insulin, so hexokinase will not be significantly active except when insulin secretion is high.
What is the role of hexokinase in the liver?
Hexokinase acts at a constant rate in the liver, and so serves to maintain a constant supply of glucose 6-phosphate for the liver’s own metabolic needs.
What effect do you think changes in the plasma concentration of glucose will
have on the rate of formation of glucose 6-phosphate catalysed by glucokinase
in the liver?
The Km of glucokinase is higher than fasting concentrations of glucose, so it only has significant activity when there is a great deal of glucose coming into the liver from the gut after a meal. The more glucose there is available, the faster glucokinase will form glucose 6-phosphate.
What do you think is the importance of glucokinase in the liver?
The increased glucose 6-phosphate formed by glucokinase as glucose entering the liver increases is used for synthesis of glycogen and fatty acids.
Glucokinase provides the first stage in controlling the amount of glucose entering the peripheral circulation . The concentration of glucose in the hepatic portal vein may be 20 mmol/L or higher; in the blood leaving the liver (and hence in the peripheral circulation) the concentration of glucose is normally no more than about 10 mmol /L.
Froguel and coworkers (1993) reported studies of the glucokinase gene in a number of families affected by MODY, and also in unaffected families. They published a list of 16 variants of the glucokinase gene. All their patients with MODY had an abnormality of the glucokinase gene:
| codon | nucleotide change |
amino acid change |
effect |
| 4 | GAC to AAC |
? |
none |
| 10 | GCC to GCT |
? |
none |
| 70 | GAA to AAA |
? |
MODY |
| 98 | CAG to TAG |
? |
MODY |
| 116 | ACC to ACT |
? |
none |
| 175 | GGA to AGA |
? |
MODY |
| 182 | GTG to ATG |
? |
MODY |
| 186 | CGA to TGA |
? |
MODY |
| 203 | GTG to GCG |
? |
MODY |
| 228 | ACG to ATG |
? |
MODY |
| 261 | GGG to AGG |
? |
MODY |
| 279 | GAG to TAG |
? |
MODY |
| 300 | GAG to AAG |
? |
MODY |
| 300 | GAG to CAG |
? |
MODY |
| 309 | CTC to CCC |
? |
MODY |
| 414 | AAG to GAG |
? |
MODY |
From data reported by Froguel P et al. 1993, ‘Familial hyperglycaemia
due to mutations in glucokinase: definition of a subtype of diabetes mellitus’,
New England Journal of Medicine, 328: 697-702
Click here for a copy of the genetic code to work out the amino acid changes
Why do you think the mutations affecting codons 4, 10, and 116 had no effect ?
| codon | nucleotide change |
amino acid change |
effect |
| 4 | GAC to AAC |
aspartate to asparagine (acidic to neutral) | none |
| 10 | GCC to GCT |
alanine to alanine (no change) | none |
| 70 | GAA to AAA |
glutamate to lysine (acidic to basic) | MODY |
| 98 | CAG to TAG |
glutamine to STOP (premature termination of translation) | MODY |
| 116 | ACC to ACT |
threonine to threonine (no change) | none |
| 175 | GGA to AGA |
glycine to arginine (small neutral basic) | MODY |
| 182 | GTG to ATG |
valine to methionine (neutral branched chain to neutral) | MODY |
| 186 | CGA to TGA |
arginine to STOP (premature termination of translation) | MODY |
| 203 | GTG to GCG |
valine to alanine (branched chain to small neutral) | MODY |
| 228 | ACG to ATG |
threonine to methionine (hydrophilic to hydrophobic) | MODY |
| 261 | GGG to AGG |
glycine to arginine (small neutral to basic) | MODY |
| 279 | GAG to TAG |
glutamate to STOP (premature termination of translation) | MODY |
| 300 | GAG to AAG |
glutamate to lysine (acidic to basic) | MODY |
| 300 | GAG to CAG |
glutamate to glutamine (acidic to neutral) | MODY |
| 309 | CTC to CCC |
leucine to proline (branched chain to Pro which breaks alpha helix) | MODY |
| 414 | AAG to GAG |
lysine to glutamate (basic to acidic) |
MODY |
The mutation in codon 4 is a significant change, from aspartate (which is acidic) to asparagine (which is neutral). However, this is the 4th amino acid from the amino terminal of the protein, and is unlikely to be important for the folding of the protein to form the active site.
The mutations in codons 10 and 116 are silent, since they do not cause any change in the amino acid.
All of the other mutations lead to either premature termination of translation, and hence no active enzyme, or a radical change in the amino acid encoded, which may reduce the activity of the enzyme considerably, by reducing its Vmax or increasing its Km. Mutations that affect the amino acids that make up the catalytic or substrate-binding site may lead to complete loss of activity.
What conclusions can you draw from the results in the table above?
The results tell us that all patients with MODY had defective glucokinase, and the table shows us that all but one of the people with abnormal glucokinase also had MODY. This suggests very strongly that a defect in glucokinase is the underlying cause of MODY.
Since this study by Froguel et al., more than 130 MODY-associated
mutations have been found in the glucokinase gene.
Froguel and coworkers (1993) studied the secretion of insulin in response to
glucose infusion in patients with MODY and normal control subjects. They were
given an intravenous infusion of glucose; the rate of infusion was varied so
as to maintain a constant plasma concentration of 10 mmol/L.
Their plasma concentrations of glucose and insulin were measured in the fasting
state and and after 60 minutes of glucose infusion:
plasma glucose (mmol /L) |
plasma insulin (mU /L) |
|||
patients |
control subjects |
patients |
control subjects |
|
| fasting | 7.0 ± 0.4 |
5.1 ± 0.3 |
5 ± 2 |
6 ± 2 |
| 60 in infusion | maintained at 10 mmol /L by varying
the rate of infusion |
12 ± 7 |
40 ± 11 |
|
From data reported by Froguel P et al. 1993, ‘Familial hyperglycaemia due to mutations in glucokinase: definition of a subtype of diabetes mellitus’, New England Journal of Medicine, 328: 697-702
What conclusions can you draw from this information about the probable role
of glucokinase in the beta-islet cells of the pancreas?
In the fasting state the patients were able to secrete a more or less normal
amount of insulin, although this was not sufficient to maintain a normal fasting
blood glucose concentration. This suggests that although they can synthesise
and secrete insulin (unlike type I diabetes), they cannot secrete an appropriate
amount to reduce the abnormally high fasting blood glucose. This suggests that
their problem may be an inability to secrete additional insulin in response
to glucose. (This is unlike type II diabetes, where insulin secretion in response
to glucose is normal or higher than normal, but tissues are insensitive to insulin).
In response to the glucose infusion, the patients are unable to increase their
insulin secretion appropriately. This confirms that their problem is secreting
insulin in response to increasing blood glucose.
From this information it seems most likely that the additional glucose 6-phosphate synthesised by glucokinase in response to increasing glucose entering the pancreas acts as the sensor of increased blood glucose and indirectly triggers secretion of insulin.
The patients can increase their secretion of insulin in response to glucose to some extent, but not as much as normal. This suggests that they are heterozygotes, with one normal and one abnormal gene for glucokinase. The result is about half the normal activity of glucokinase in the cells, and this is not enough to provide enough glucose 6-phosphate in response to increased glucose to stimulate full insulin secretion. Hence the dominant pattern of inheritance.
Glucose 6-phosphate is not the immediate signal for insulin secretion. Rather, it is metabolised, leading to increased formation of ATP. This leads to closure of an ATP-potassium channel, causing membrane depolarisation and opening of a voltage-gated calcium channel. The resultant influx of calcium ions leads to fusion of the insulin secretory granules with the cell membrane, and the release of insulin .
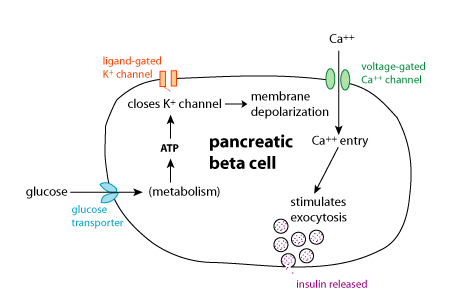
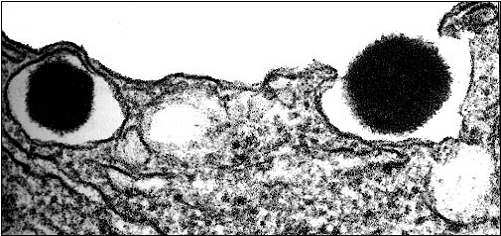
The electron micrograph below shows secretion of insulin by exocytosis from secretory granules that have fused with the cell membrane, from Alberts B, et al., Molecular Biology of the Cell (3rd Ed). Garland Publishing, New York, 1994.
There is some evidence that glucokinase is alsoexpressed in the alpha-islet cells of the pancreas, that secrete glucagon. Here the role of the additional glucose 6-phosphate produced in response to an increase in blood glucose is to inhibit the secretion of glucagon, presumably by the same mechanism as the stimulation of insulin secretion.